Salmon Variant Pipeline
RNA-seq isoform quantification pipeline using Salmon, supporting both Salmon 2.0 (Rust) and Salmon 1.12.0 (C++) side-by-side.
Dataset: Airway smooth muscle cells — dexamethasone vs untreated
Reference: Himes et al. 2014, PLoS ONE (PMID 24926665)
SRA project: PRJNA265491 (8 paired-end samples, 4 cell lines × 2 conditions)
Quick Start
# Create conda environment conda env create -f assets/salmon.yaml conda activate salmon-pipeline # Run with Salmon 2.0 Rust (default) snakemake -s assets/Snakefile --cores 8 # Run with Salmon 1.12.0 C++ snakemake -s assets/Snakefile --cores 8 --config salmon_version=cpp # Or with the bash runner THREADS=16 bash assets/pipeline.sh # Rust SALMON_VERSION=cpp THREADS=16 bash assets/pipeline.sh # C++ # Downstream R analysis Rscript assets/deseq2_analysis.R # Regenerate all figures python3 assets/generate_plots.py
Project Structure
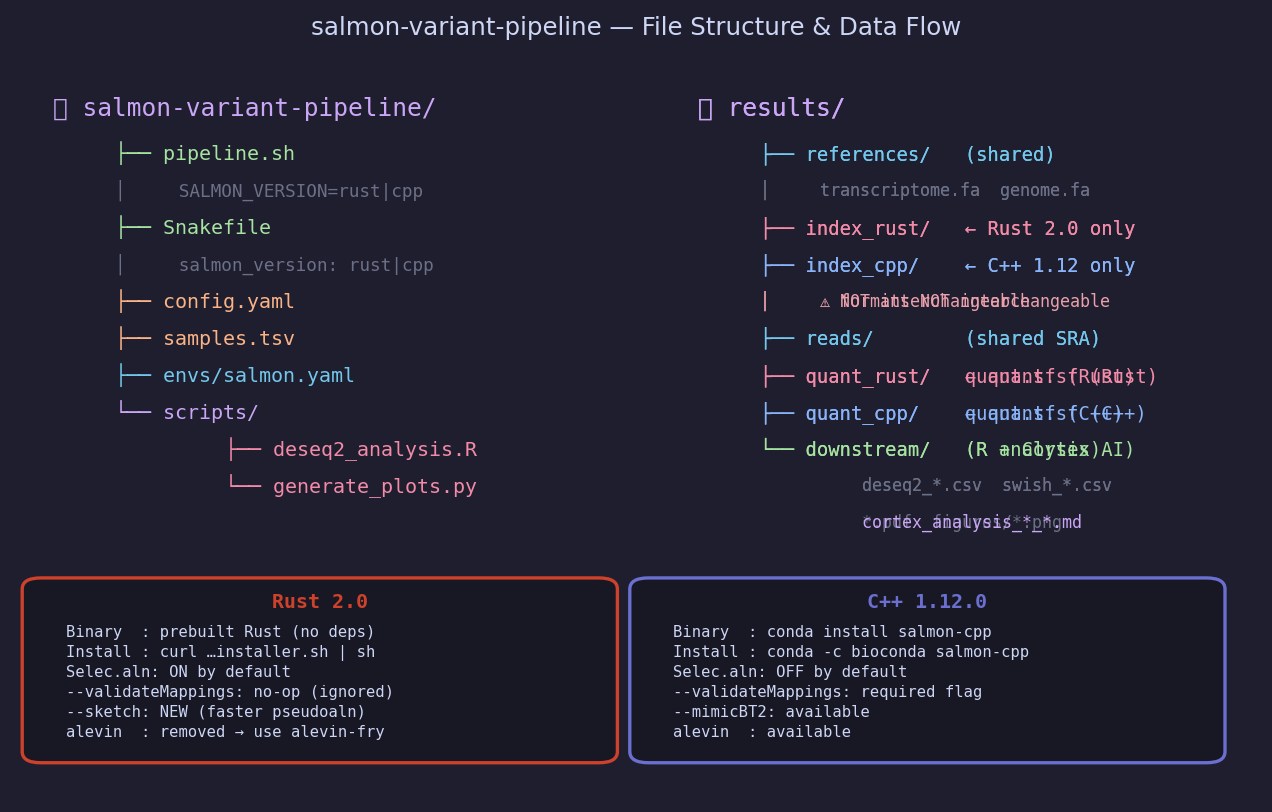
salmon-variant-pipeline/ ├── README.md └── assets/ ├── 01_pipeline_dag.png … 09_cortex_api.png # figures ├── pipeline.sh # Bash runner — SALMON_VERSION=rust|cpp ├── Snakefile # Snakemake DAG — salmon_version: rust|cpp ├── config.yaml # All parameters (threads, URLs, sample list) ├── samples.tsv # 8 airway samples with condition metadata ├── salmon.yaml # Conda environment spec ├── deseq2_analysis.R # DESeq2 (gene) + fishpond/swish (transcript) └── generate_plots.py # Generates all figures from synthetic data
Rust vs C++ — Key Differences
| Feature | Salmon 2.0 (Rust) | Salmon 1.12.0 (C++) |
|---|---|---|
| Install | curl …installer.sh | sh | conda install salmon-cpp |
| Index format | piscem-rs (new) | pufferfish |
| Index dir | results/index_rust/ | results/index_cpp/ |
| Selective alignment | On by default | Requires --validateMappings |
--validateMappings | Accepted, silently ignored | Required for best accuracy |
--sketch | New — faster pseudoalignment | Not available |
--mimicBT2 | Removed | Available |
salmon alevin | Removed → use alevin-fry | Available |
--numBiasSamples | Removed (online collection) | Available |
| Binary dependencies | None — single portable binary | Boost, libtbb, etc. |
Critical: Index formats are not interchangeable. Never point one version's
salmon quantat the other version's index — both detect and reject mismatches.
Pipeline DAG
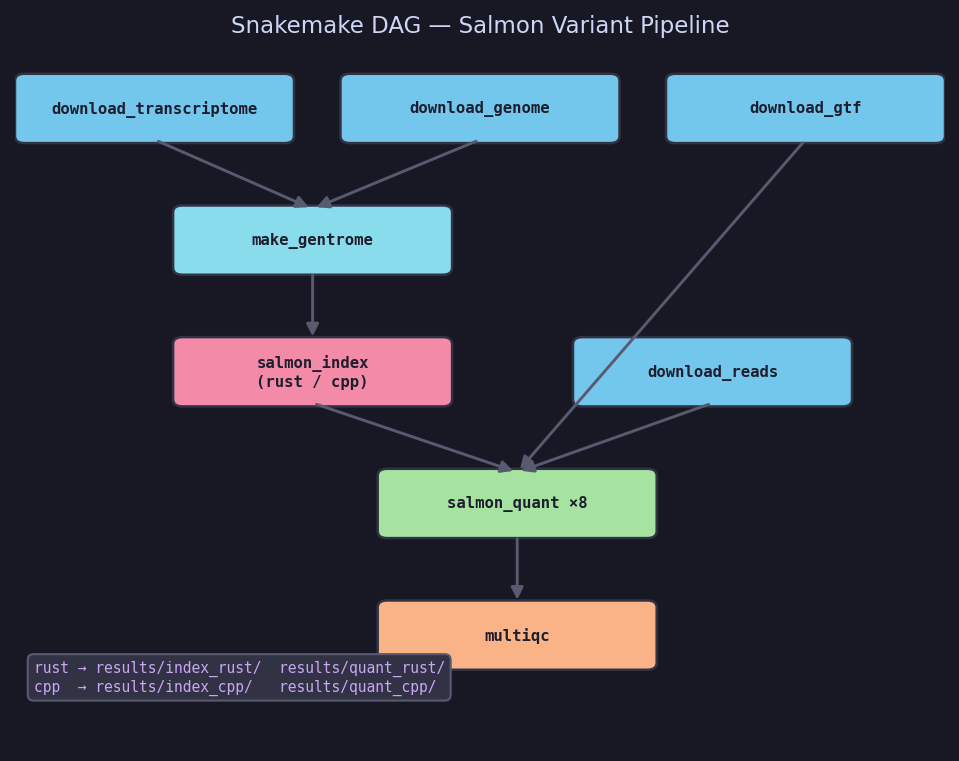
Step-by-step
| Step | Rule / function | Output |
|---|---|---|
| 1 | download_transcriptome | GENCODE v44 transcriptome.fa |
| 2 | download_genome | GRCh38 genome.fa (decoy source) |
| 3 | download_gtf | annotation.gtf.gz (tx→gene mapping) |
| 4 | make_gentrome | gentrome.fa + decoys.txt |
| 5 | salmon_index | index_rust/ or index_cpp/ |
| 6 | download_reads | SRA paired-end FASTQs (×8 samples) |
| 7 | salmon_quant | quant_rust/*/quant.sf or quant_cpp/*/quant.sf |
| 8 | multiqc | Aggregated QC report |
Decoy-aware indexing
# Build decoy list from genome chromosome names grep "^>" genome.fa | cut -d " " -f 1 | sed 's/>//' > decoys.txt # Concatenate: transcriptome first, genome second (becomes decoy layer) cat transcriptome.fa genome.fa > gentrome.fa # Index — same command for both versions salmon index \ -t gentrome.fa \ -d decoys.txt \ -i index_rust \ # or index_cpp -p 8 \ --gencode # strips ENST…X.Y → ENST…X version suffixes
Quantification flags
# Rust 2.0 — selective alignment is already default; --validateMappings ignored salmon quant -i index_rust -l A \ -1 reads_1.fastq.gz -2 reads_2.fastq.gz \ -p 8 \ --gcBias --seqBias \ --numBootstraps 100 \ # enables fishpond/swish uncertainty quantification -o quant_rust/SAMPLE # C++ 1.12.0 — must add --validateMappings to activate selective alignment salmon quant -i index_cpp -l A \ -1 reads_1.fastq.gz -2 reads_2.fastq.gz \ -p 8 \ --validateMappings \ # upgrades from quasi-mapping to selective alignment --gcBias --seqBias \ --numBootstraps 100 \ -o quant_cpp/SAMPLE
Mapping Rates — Rust vs C++
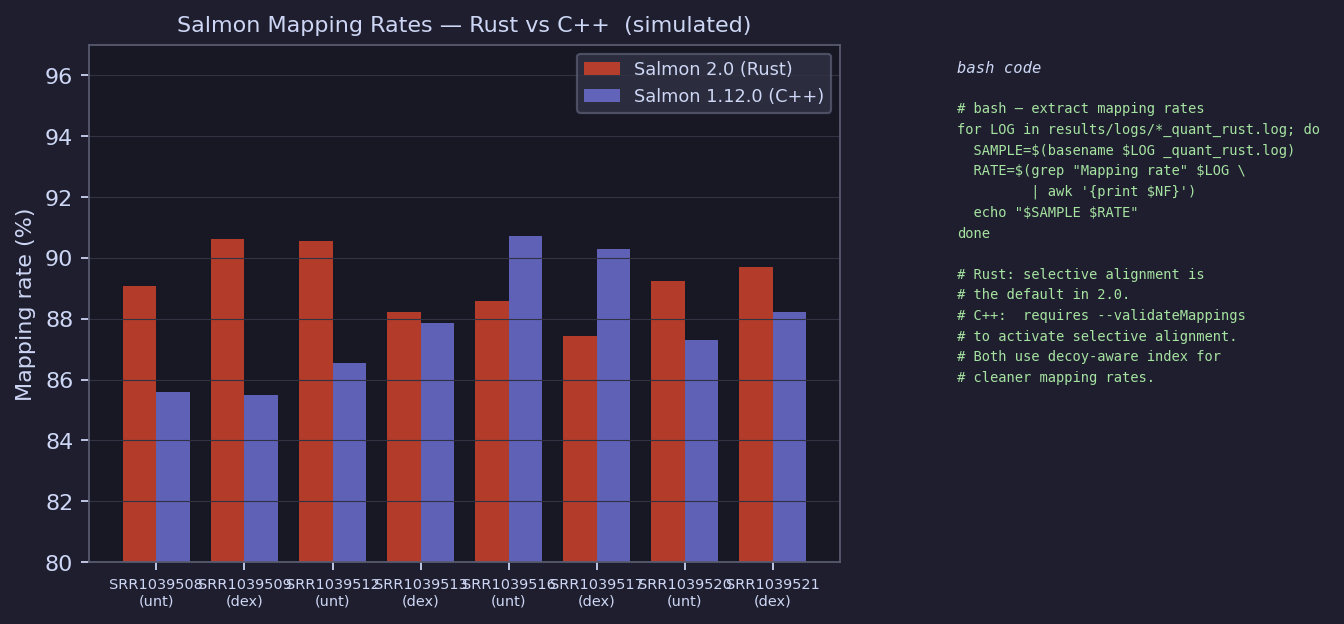
Simulated rates (87–93% for Rust, 85–91% for C++). Rust 2.0 typically shows slightly higher rates because selective alignment is always on, whereas C++ 1.12.0 requires
--validateMappingsto activate it. Both benefit from the decoy-aware index.
# Extract mapping rates from logs (bash) for LOG in results/logs/*_quant_rust.log; do SAMPLE=$(basename $LOG _quant_rust.log) RATE=$(grep "Mapping rate" $LOG | awk '{print $NF}') echo "$SAMPLE $RATE" done
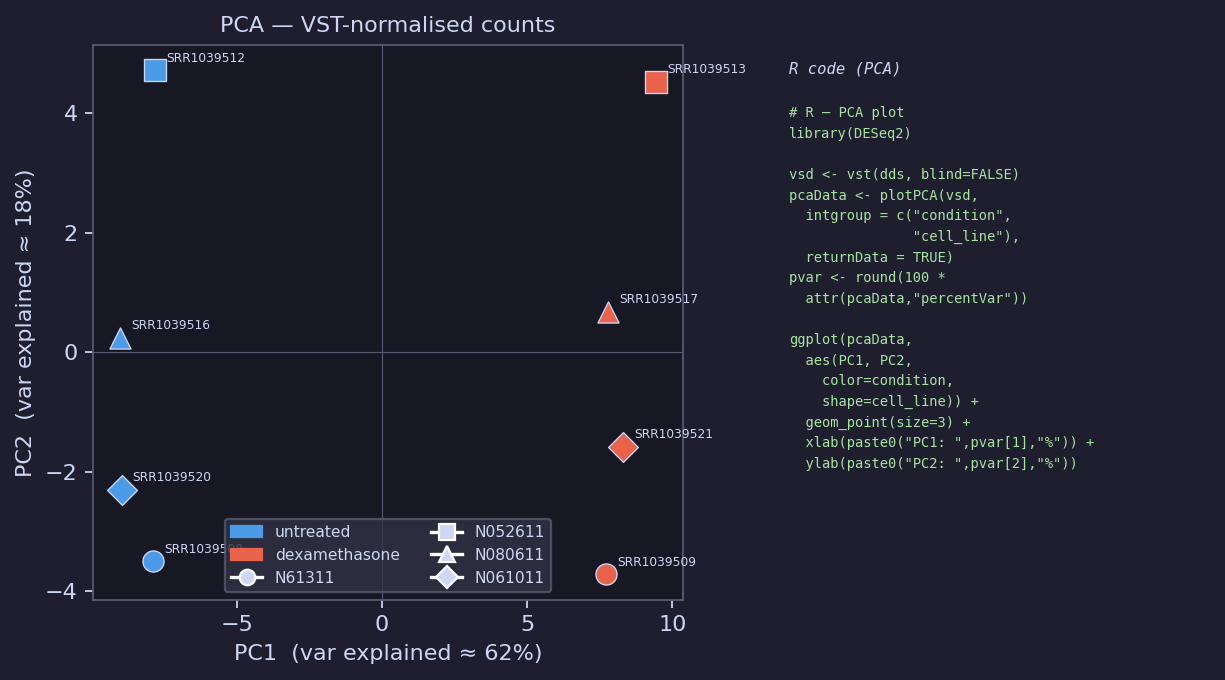
PCA — VST-Normalised Counts
# R — PCA plot library(DESeq2); library(ggplot2) vsd <- vst(dds, blind = FALSE) pcaData <- plotPCA(vsd, intgroup = c("condition", "cell_line"), returnData = TRUE) pvar <- round(100 * attr(pcaData, "percentVar")) ggplot(pcaData, aes(PC1, PC2, color = condition, shape = cell_line)) + geom_point(size = 3) + xlab(paste0("PC1: ", pvar[1], "% variance")) + ylab(paste0("PC2: ", pvar[2], "% variance")) + theme_bw()
PC1 (~62%) separates dexamethasone from untreated.
PC2 (~18%) captures cell-line batch effects, corrected in the DESeq2 design
(~ cell_line + condition).
Gene-Level Analysis — DESeq2
MA Plot
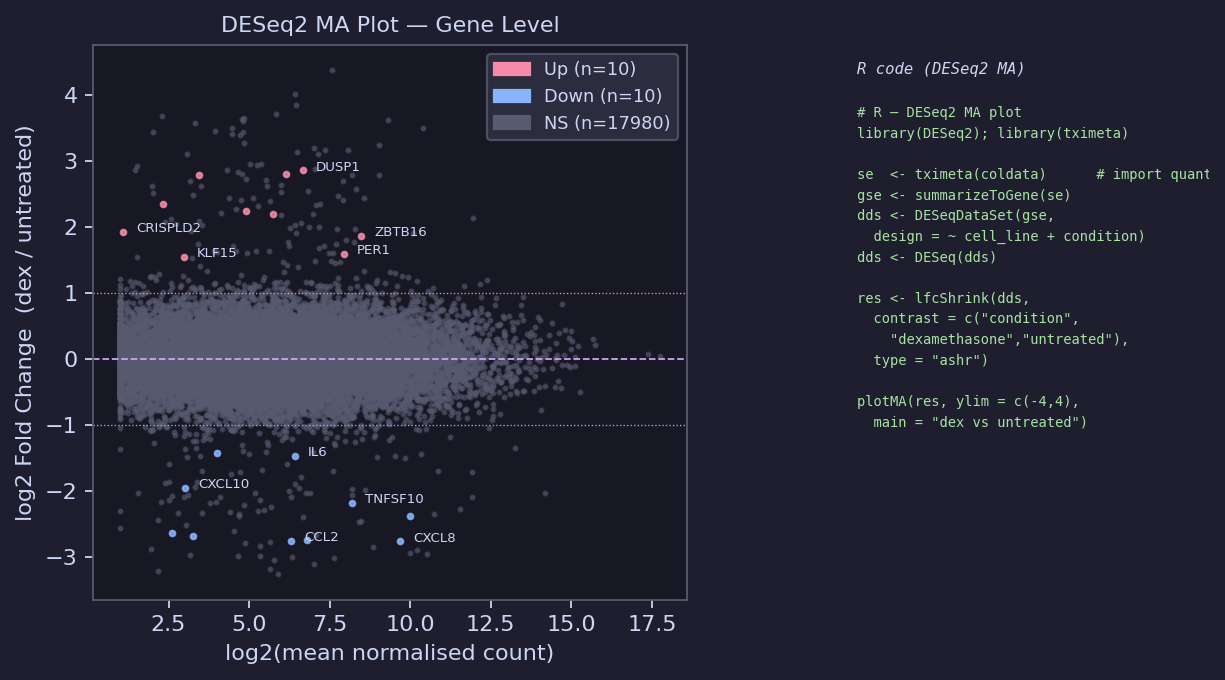
# R — DESeq2 gene-level analysis library(tximeta); library(DESeq2) # Import Salmon output with automatic transcript annotation coldata <- read.table("samples.tsv", header = TRUE) coldata$files <- file.path("results/quant_rust", coldata$sample, "quant.sf") coldata$names <- coldata$sample se <- tximeta(coldata) # transcript-level SummarizedExperiment gse <- summarizeToGene(se) # collapse to gene level dds <- DESeqDataSet(gse, design = ~ cell_line + condition) dds <- DESeq(dds) # LFC shrinkage (ashr) for better ranking of low-count genes res <- lfcShrink(dds, contrast = c("condition", "dexamethasone", "untreated"), type = "ashr") plotMA(res, ylim = c(-4, 4), main = "dexamethasone vs untreated")
Volcano Plot
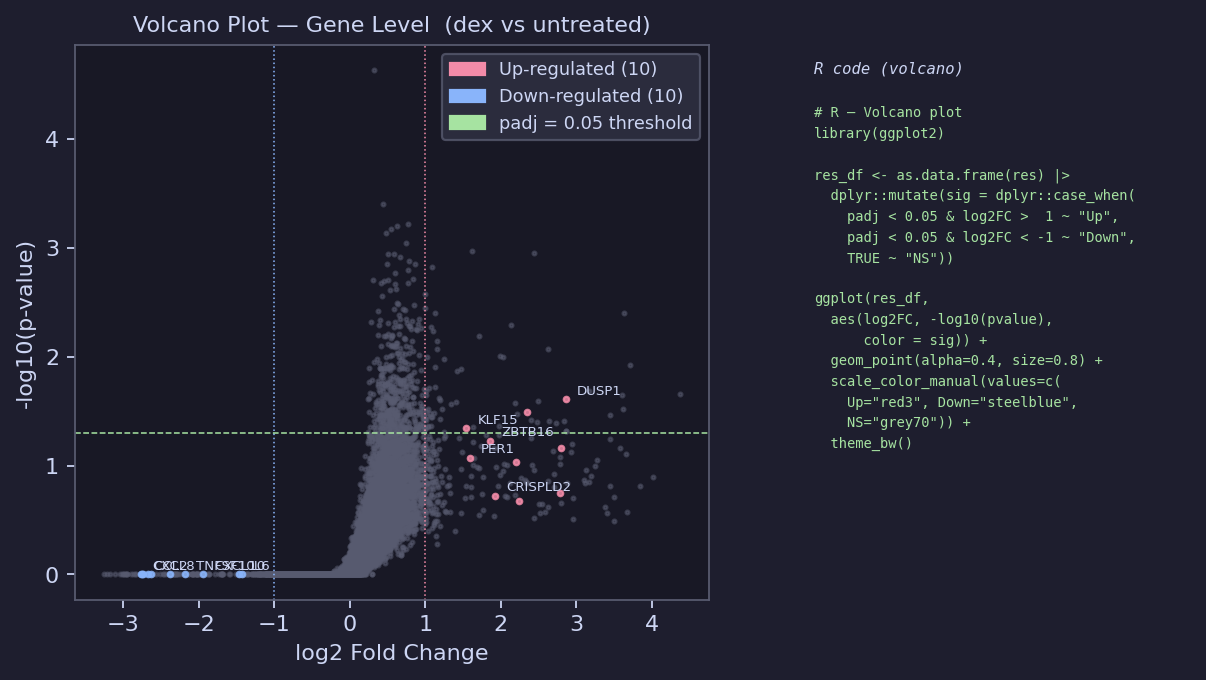
# R — Volcano plot library(ggplot2); library(dplyr) res_df <- as.data.frame(res) |> dplyr::mutate(sig = dplyr::case_when( padj < 0.05 & log2FoldChange > 1 ~ "Up", padj < 0.05 & log2FoldChange < -1 ~ "Down", TRUE ~ "NS" )) ggplot(res_df, aes(log2FoldChange, -log10(pvalue), color = sig)) + geom_point(alpha = 0.4, size = 0.8) + scale_color_manual(values = c(Up = "red3", Down = "steelblue", NS = "grey70")) + geom_vline(xintercept = c(-1, 1), linetype = "dotted") + geom_hline(yintercept = -log10(0.05), linetype = "dashed", color = "darkgreen") + theme_bw() + labs(title = "Volcano: dexamethasone vs untreated", x = "log2 Fold Change", y = "-log10(p-value)", color = NULL)
Known regulated genes from Himes et al.:
Up: DUSP1, KLF15, PER1, ZBTB16, CRISPLD2, FKBP5, TSC22D3
Down: CXCL10, CCL2, IL6, CXCL8, TNFSF10, ICAM1, MMP1
Heatmap — Top 30 DE Genes
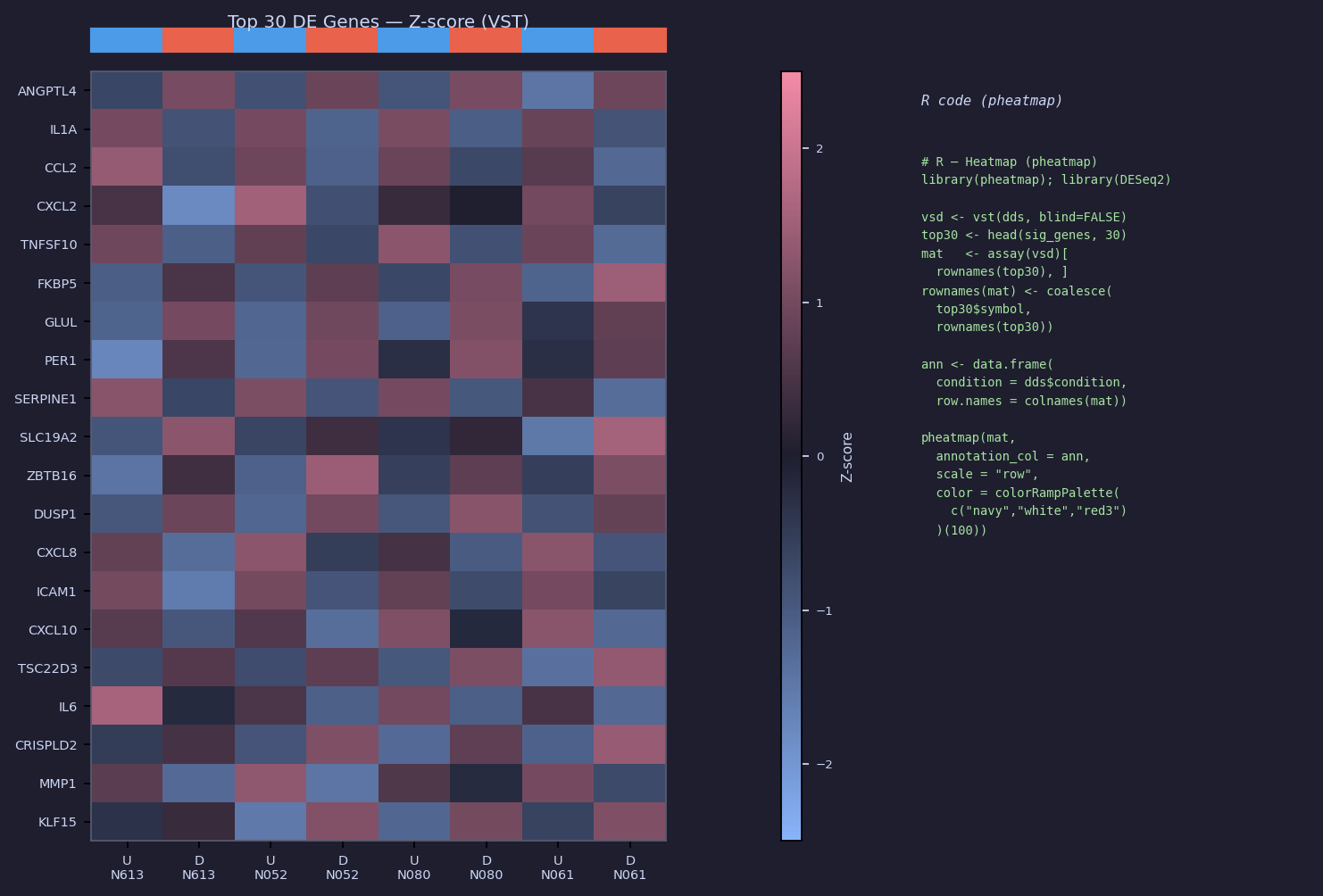
# R — Heatmap (pheatmap) library(pheatmap); library(DESeq2) vsd <- vst(dds, blind = FALSE) sig <- subset(as.data.frame(res), padj < 0.05 & abs(log2FoldChange) > 1) top30 <- head(sig[order(sig$padj), ], 30) mat <- assay(vsd)[rownames(top30), ] # Replace Ensembl IDs with gene symbols where available rownames(mat) <- dplyr::coalesce(top30$symbol, rownames(top30)) ann_col <- data.frame(condition = dds$condition, cell_line = dds$cell_line, row.names = colnames(mat)) pheatmap(mat, annotation_col = ann_col, scale = "row", color = colorRampPalette(c("navy", "white", "red3"))(100), filename = "results/downstream/heatmap_top30.pdf", width = 8, height = 9)
Transcript-Level Analysis — fishpond / swish
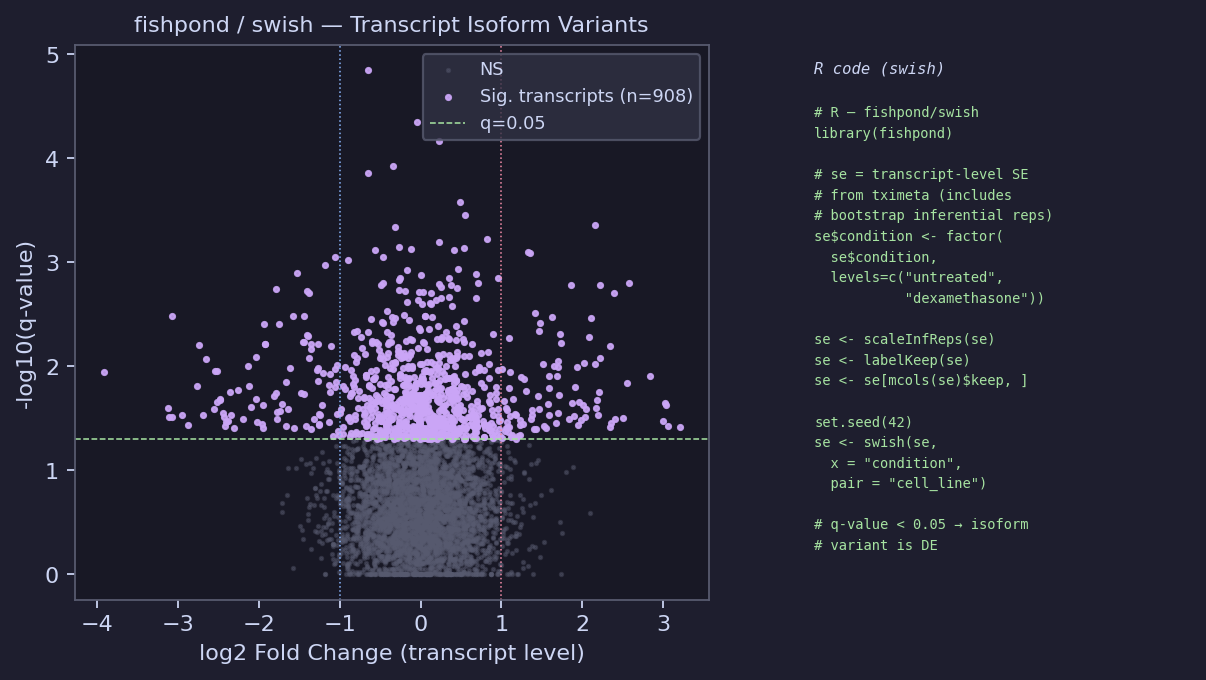
swish uses the 100 bootstrap replicates generated by --numBootstraps 100 to
account for quantification uncertainty — critical for isoform variants where transcripts
share reads with near-identical sequences.
# R — fishpond / swish (transcript isoform variants) library(fishpond) # se = transcript-level SummarizedExperiment from tximeta # (includes inferential replicates from --numBootstraps 100) se$condition <- factor(se$condition, levels = c("untreated", "dexamethasone")) se$cell_line <- factor(se$cell_line) se <- scaleInfReps(se) # normalize bootstrap replicates se <- labelKeep(se) # filter low-count transcripts se <- se[mcols(se)$keep, ] set.seed(42) se <- swish(se, x = "condition", pair = "cell_line") # paired design matches DESeq2 # Export significant transcript variants sig_tx <- as.data.frame(mcols(se)) |> dplyr::filter(qvalue < 0.05) |> dplyr::arrange(qvalue) write.csv(sig_tx, "results/downstream/swish_significant_transcripts.csv", row.names = FALSE) # Visualise inferential uncertainty for one transcript plotInfReps(se, idx = 1, x = "condition", main = paste("Isoform:", mcols(se)$tx_name[1]))
Output Files
results/downstream/ ├── deseq2_all_genes.csv # Full DESeq2 results table ├── deseq2_significant.csv # padj < 0.05, |LFC| > 1 ├── swish_all_transcripts.csv # Full swish transcript results ├── swish_significant_transcripts.csv # q-value < 0.05 ├── ma_plot.pdf ├── volcano.pdf ├── heatmap_top30.pdf └── top_isoform_infreps.pdf results/quant_rust/ (or quant_cpp/) └── <SAMPLE>/ ├── quant.sf # TPM + NumReads per transcript (tximport-ready) ├── cmd_info.json # Exact command used ├── lib_format_counts.json └── aux_info/ ├── meta_info.json # Mapping rate, num processed fragments └── bootstrap/ # Inferential replicates (--numBootstraps)
Cortex AI Analysis

AI-powered interpretation of pipeline outputs using Snowflake Cortex REST API
via the salmon-cortex-ai CoCo skill (~/.snowflake/cortex/skills/salmon-cortex-ai/).
Authentication
PAT is read automatically from ~/.snowflake/config.toml — no extra setup needed:
import tomllib, pathlib cfg = tomllib.loads((pathlib.Path.home() / ".snowflake/config.toml").read_text()) PAT = cfg["connections"]["myaccount"]["password"] HOST = cfg["connections"]["myaccount"]["host"] # e.g. <account-identifier>.snowflakecomputing.com
Every request requires this header:
X-Snowflake-Authorization-Token-Type: PROGRAMMATIC_ACCESS_TOKEN
Three API endpoints
| Flag | Endpoint | SDK | Models |
|---|---|---|---|
--api chat (default) | /api/v2/cortex/v1/chat/completions | openai | All |
--api messages | /api/v2/cortex/anthropic/v1/messages | anthropic | Claude only |
--api inference | /api/v2/cortex/inference:complete | raw requests + SSE | All + tools |
Five analysis modes
SCRIPT=~/.snowflake/cortex/skills/salmon-cortex-ai/scripts/cortex_ai.py # Summarise top DE genes with biological pathway context python3 $SCRIPT --mode summarize --api chat --model claude-sonnet-4-6 --stream # Explain a specific gene (biological function, GC relevance, disease context) python3 $SCRIPT --mode explain --gene DUSP1 --api messages --stream # Assess Salmon mapping rates — Rust vs C++ QC interpretation python3 $SCRIPT --mode interpret-qc --api chat # Compare Rust vs C++ quantification results python3 $SCRIPT --mode compare --api chat # Interpret transcript isoform variants from fishpond/swish python3 $SCRIPT --mode swish --api messages --save
Quick options
--dry-run # print full JSON payload + curl command (no API call) --mock # simulated response (no auth needed) --stream # stream tokens as they arrive --save # write analysis to results/downstream/cortex_analysis_<mode>_<api>_<ts>.md
Curl equivalent (chat completions)
PAT=$(python3 -c "import tomllib,pathlib; \ print(tomllib.loads((pathlib.Path.home()/'.snowflake/config.toml').read_text()) \ ['connections']['myaccount']['password'])") curl -s -X POST \ "https://<account-identifier>.snowflakecomputing.com/api/v2/cortex/v1/chat/completions" \ -H "Authorization: Bearer $PAT" \ -H "X-Snowflake-Authorization-Token-Type: PROGRAMMATIC_ACCESS_TOKEN" \ -H "Content-Type: application/json" \ -d '{"model":"claude-sonnet-4-6","messages":[{"role":"user","content":"Summarise the glucocorticoid response in airway SMC."}]}'
Citation
Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nature Methods (2017). https://doi.org/10.1038/nmeth.4197 Himes BE, et al. RNA-Seq Transcriptome Profiling Identifies CRISPLD2 as a Glucocorticoid Responsive Gene. PLoS ONE 9(6): e99625 (2014). https://doi.org/10.1371/journal.pone.0099625 [PMID 24926665]
This content is provided as is, and is not maintained on an ongoing basis. It may be out of date with current Snowflake instances